Industrie pharmaceutique et réglementation

L’industrie pharmaceutique est très réglementée : quelle est l’origine de cette réglementation ?
L’industrie pharmaceutique est très réglementée : quelle est l’origine de cette réglementation ?
La constitution de l’Organisation mondiale de la santé de 1946 indique que : « La possession du meilleur état de santé qu’il est capable d’atteindre constitue l’un des droits fondamentaux de tout être humain ». Cette déclaration est symptomatique de l’importance que l’on accorde aujourd’hui à la santé. Le préambule de la Constitution de 1946 qui a toujours valeur constitutionnelle sous la Ve République consacre en France un droit fondamental à la protection de la santé.
Depuis dix ans, la sécurité sanitaire s’est imposée comme une composante essentielle de la politique de santé française. La sécurité sanitaire repose fondamentalement sur la notion de rapport bénéfices/risques : un produit de santé ne doit être diffusé que si les bénéfices qu’il apporte au patient l’emportent sur les risques qu’il comporte. La plupart des pays ont développé des réglementations du même type. Les règles pharmaceutiques sont d’ailleurs désormais largement internationales même si leur application reste très inégale.
Quelle est la situation française en ce qui concerne ces règles ?
La réglementation française du médicament est particulièrement développée et rigoureuse. Cela s’explique d’abord par les drames sanitaires que notre pays comme d’autres, peut-être plus que d’autres, a connus.
Les affaires du Stalinon, de la Thalidomide et du Distilbène1, médicaments dangereux dont l’évaluation initiale et la surveillance n’avaient pas été assez strictes, ont malheureusement fait prendre conscience tôt des conséquences sanitaires possibles des carences ou des fautes des systèmes de santé publique. Des procédures de mise sur le marché, puis de pharmacovigilance ont ainsi été définies et mises en œuvre. Les drames de santé publique récents, en particulier le drame du sang contaminé et des hormones de croissance, qui ont touché des milliers de personnes, ont renforcé la prise de conscience des risques liés aux produits de santé et conduit à un renforcement sans précédent des règles.
Par ailleurs, en tant que produits industriels les produits de santé sont entrés dans le champ du traité de Rome bien avant l’inscription de la santé publique dans les compétences de la Communauté par les traités de Maastricht et d’Amsterdam. Soumis à la libre circulation, ils ont également été soumis à des règles communes de sécurité.
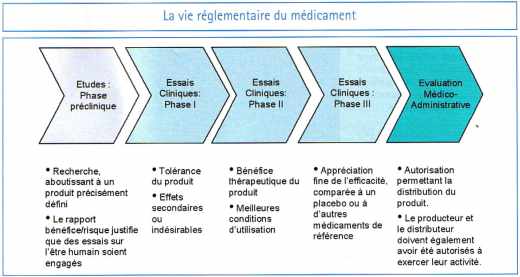
C’est ainsi qu’ont été mis en place un encadrement réglementaire strict et développé un corpus de bonnes pratiques. Le dispositif est particulièrement complet. Des essais cliniques à l’autorisation de mise sur le marché d’un médicament, de la pharmacovigilance au contrôle en laboratoire ou par voie d’inspection, le système vise à connaître en permanence le rapport bénéfices/risques du produit et à réagir en cas d’évolution défavorable de celui-ci.
Quel est le système d’autorisation de mise sur le marché ?
Un système d’autorisation a été créé dans chaque pays pour le médicament, qui est appelé en France Autorisation de mise sur le marché (AMM). L’octroi ou non d’une AMM est une décision prise à l’issue d’un processus d’évaluation dont le but est, conformément au principe qui sous-tend la réglementation, de mesurer a priori le rapport bénéfices/risques du médicament.
… dans le cas de la France
L’évaluation se fonde en particulier sur les données obtenues par le laboratoire demandant l’AMM au cours des études cliniques (voir encadré). Depuis 1993, il s’agit d’une double évaluation : évaluation interne par les scientifiques de l’Agence française de sécurité sanitaire des produits de santé (Afssaps, ex-Agence du médicament) et externe par une commission d’AMM rattachée à l’Afssaps. Le principe est d’avoir à la fois une évaluation par des spécialistes du domaine indépendants de l’Afssaps, et une évaluation par des experts internes qui garantissent la continuité de l’évaluation et des dossiers.
Au terme de l’évaluation, la commission d’AMM émet un avis, qui aboutit à une décision du directeur général de l’Afssaps, habilité par le code de la santé publique depuis la loi du 4 janvier 1993 à délivrer l’AMM au nom de l’État. Auparavant l’AMM était délivrée par le ministre de la Santé.
… et en Europe
En Europe, deux procédures coexistent.
Tout d’abord, pour une grande partie des produits, une procédure de reconnaissance mutuelle permet qu’une AMM obtenue dans un État membre soit reconnue dans les autres États membres de l’Union, ce qui donne lieu à la délivrance d’une autorisation de mise sur le marché par chaque État concerné.
Par ailleurs, les produits innovants comme les produits issus des biotechnologies sont obligatoirement évalués par l’Agence européenne pour l’évaluation des médicaments (EMEA). Cette évaluation est effectuée par un panel d’experts provenant des agences nationales, ce qui permet de prendre en compte les points de vue des divers pays. C’est le commissaire européen, sur avis de l’EMEA, qui délivre une autorisation de mise sur le marché valable dans l’ensemble du territoire de l’Union.
Ce système d’agence tête de réseau des agences nationales est assez efficace, car il permet d’avoir des agences près du terrain, tout en gérant les intérêts communs à un niveau communautaire. Une FDA à l’européenne, avec une agence unique, fonctionnerait certainement moins bien du fait de sa lourdeur par rapport au réseau actuel, tant pour la qualité de l’expertise que pour ce qui est essentiel, la réactivité en cas d’alerte sanitaire.
… et dans le monde
Outre le système européen, les deux autres grands systèmes dans le monde sont la Food and Drug Administration (FDA) américaine et le ministère de la Santé japonais (MHLW).
Chaque système a ses propres caractéristiques et reste indépendant. L’évaluation est faite de façon différente par chaque agence du fait de ses principes d’organisation mais aussi de situations épidémiologiques diverses justifiant des évaluations adaptées d’un pays à l’autre.
Les conférences International Conference for Harmonization (ICH) ont pour but d’harmoniser les critères d’évaluation et de jugement partout dans le monde, avec des normes communes, mais il s’agit d’un processus lent et de véritables différences d’approche demeurent entre les différents systèmes.
Malgré ces différences, les informations sont très internationalisées et toute alerte sur un sujet sérieux est très rapidement mondiale.
Vous avez mentionné que la réglementation ne s’arrête pas à l’autorisation sur le marché : le médicament est-il encore évalué une fois sur le marché ?
Il s’agit d’une composante cruciale du système : la pharmacovigilance, réévaluation permanente des médicaments, de leur rapport bénéfices/ risques, au regard des informations nouvelles sur les risques. C’est aussi important que l’évaluation initiale.
Cette réévaluation exige de prendre en compte en permanence l’état des connaissances, en constante évolution, sur les effets indésirables, non seulement des molécules elles-mêmes, mais également d’éléments extérieurs au champ du médicament, tels que la toxicité des composants, l’observance des traitements par les patients, les usages détournés des médicaments, etc.
Toute information pouvant avoir une incidence sanitaire sérieuse, une veille scientifique très élaborée est nécessaire. Dans ce cadre, la mise en commun au niveau européen et mondial des données est précieuse, car elle augmente considérablement les chances que rien n’échappe aux mailles du filet.
Tout problème rapporté après la mise sur le marché peut donner lieu à des mesures de rappels de lots, de suspension voire de retrait d’AMM2.
C’est donc au prix de ces mesures drastiques, à toutes les étapes du développement et de la vie du médicament, qu’un niveau satisfaisant de sécurité sanitaire est assuré.
Le coût des médicaments n’est pas supporté directement par les consommateurs : comment sont financées les dépenses de santé ?
Le financement des dépenses de santé est très variable selon les pays. Dans les pays dans lesquels la dépense est socialisée, elle l’est en général soit à travers des assurances privées, une sécurité sociale ou une combinaison des deux. Dans certains pays, le prix des services de santé comme leurs conditions de remboursement sont fixés par le gouvernement. Enfin, la plupart des médicaments doivent être prescrits. Il ne s’agit donc pas au sens strict d’une économie de marché. Enfin chaque pays européen met en place un système propre permettant de maîtriser les dépenses de santé. Les politiques de prix et de remboursement restent donc très nationales.
En France, la réglementation de la Sécurité sociale comprend à la fois la régulation sur le prix et sur le remboursement.
Qu’en est-il du remboursement ?
Les choix faits en matière de sécurité sociale sont en principe faits comme pour la sécurité sanitaire, à l’issue d’une évaluation, utilisant en particulier une méthodologie d’évaluation pharmacoéconomique afin de se fonder sur des critères les plus scientifiques et objectifs possibles.

Art ou Biologie ? Visualisation de la plaque dentaire. © INSERM, PHOTO KEREBEL B.
Cependant, au-delà des qualités intrinsèques du produit et de l’évaluation pharmacoéconomique, les choix faits en matière de sécurité sociale sont dépendants de la politique de santé : c’est le « Service médical rendu » (SMR), mais également « l’intérêt pour la santé publique » d’un médicament qui définissent les conditions de son remboursement.
Ce second critère est important : on peut ainsi choisir de rembourser des médicaments de SMR faible, s’ils peuvent contribuer au succès d’une politique de santé publique.
C’est une commission d’experts dépendant de l’Afssaps, la Commission de la transparence, qui émet un avis sur le remboursement et le taux de remboursement du produit.
Là encore, l’évaluation peut être remise en cause périodiquement et l’actualité récente nous le rappelle : le plan gouvernemental de 2001 prévoyait la réduction des prix ou du taux de remboursement de médicaments à SMR faible, mais le Conseil d’État, saisi par certains laboratoires, a jugé insuffisamment motivés les avis rendus par la Commission de la transparence et annulé les décisions prises.
Et comment sont arrêtés les prix ?
Depuis la fin des années 1990, le Comité économique des produits de santé (CEPS), collège de hauts fonctionnaires et de représentants de l’assurance maladie, négocie le prix des médicaments avec les laboratoires. Si les négociations aboutissent, le prix est fixé, en général, dans le cadre d’une convention conclue entre l’État et le laboratoire, signée par le président du Comité – c’est en pratique ce qui arrive dans la plupart des cas.
Cette phase de négociation de prix induit des délais, souvent dénoncés, dans la mise à disposition des patients des médicaments nouveaux et remboursés en pharmacie par rapport à la Grande-Bretagne, l’Allemagne ou les États-Unis. Cela tend à se réduire. On peut ajouter, et c’est essentiel en termes de santé publique, que les produits innovants sont au contraire très tôt disponibles à l’hôpital. Enfin, pour les produits très innovants, une procédure de « dépôt de prix » a donc été instaurée en 2003, qui devrait permettre d’accélérer sensiblement la procédure pour ces produits.
La libre circulation des produits en Europe s’accommode mal de la disparité des législations et partant des prix des médicaments. Il en résulte un problème majeur, celui des importations parallèles : des intermédiaires achètent les médicaments dans les pays où ils sont moins chers pour les revendre ailleurs en Europe, sans que les systèmes de protection sociale bénéficient de ces « rabais ».
L’une des solutions consisterait à mettre en place un prix unique en Europe, quitte à ce que chaque système récupère une « partie des prix » par des négociations dans le cadre de conventions entre les laboratoires et les divers systèmes de sécurité sociale.
Quels sont, en conclusion, les enjeux auxquels doit faire face la réglementation ?
La difficulté, dans des sociétés complexes et réglementées, que je qualifie aussi de « sociétés fragiles » est de trouver un juste équilibre entre la nécessaire prise de risque qui permet de progresser, de bénéficier de bénéfices thérapeutiques majeurs et la garantie de la sécurité sanitaire.
Pour cela, au-delà des mesures de réglementation, l’exercice de ses responsabilités par chacun des acteurs est essentiel. De l’équilibre entre réglementation et responsabilité individuelle dépendent l’équilibre et le bon fonctionnement du système.
Par ailleurs, la question du financement reste posée et là encore l’État doit trouver les leviers qui permettent de mettre en œuvre une politique de santé publique qui prenne en compte le besoin de maîtrise des dépenses de santé.
___________________________________________
1 - En France, environ 80 000 filles auraient été exposées au Distilbène pendant la grossesse de leur mère et 20 à 25 % d’entre elles pourraient être affectées par des problèmes d’infertilité.
2 - Un exemple récent (juin 2003) est celui du Pilosuryl®, retiré de la vente après que trois cas graves d’insuffisance rénale aiguë et de coma ont été rapportés lors de prises excessives du médicament.
Articles similaires :
 L’impact des biotechnologies sur la R & D pharmaceutique
L’impact des biotechnologies sur la R & D pharmaceutique
 L’impact de la génomique dans la R & D pharmaceutique : une révolution
Impact du digital sur les supply chains : l’exemple de l’industrie pharmaceutique
L’impact de la génomique dans la R & D pharmaceutique : une révolution
Impact du digital sur les supply chains : l’exemple de l’industrie pharmaceutique
 La révolution biotechnologique est en marche !
Une success-story qui se poursuit !
La révolution biotechnologique est en marche !
Une success-story qui se poursuit !